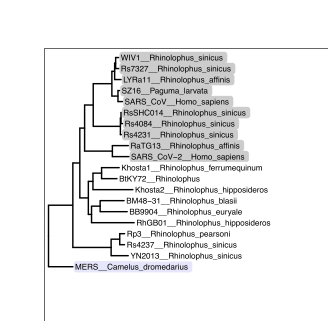
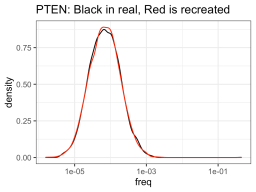
So I had collected this data over 10 months ago, when I made an initial foray into trying the fluorogen activating peptides dL5 in the presence of Malachite Green, a dirt cheap small molecule (You can use it to try to kill fungus eating at your fish in your aquarium). A recent email reminded me about fluorogen activating peptides again, so I went back and looked at this data again, and it really isn’t bad.



In summary. 1) Works well, giving ~ 100-fold increase to fluorescence from background when highly expressed. 2) Probably want to do at least 500 ng/mL or perhaps even 1ug/mL to improve signal over background. 3) Still get specific signal after washout, but you may as well leave it on and get max signal.